Hightlights:
-
-
Endogenous type I-F CRISPR-Cas is repurposed for genome editing in MDR P. aeruginosa
-
-
Mutant lines reveal key MDR determinants and extensive synergy in a clinical isolate
-
-
Clinical strain PA154197 displays collateral sensitivity to small cationic peptidomimetics
-
-
Small cationic peptidomimetics sensitize PA154197 cells to antibiotics
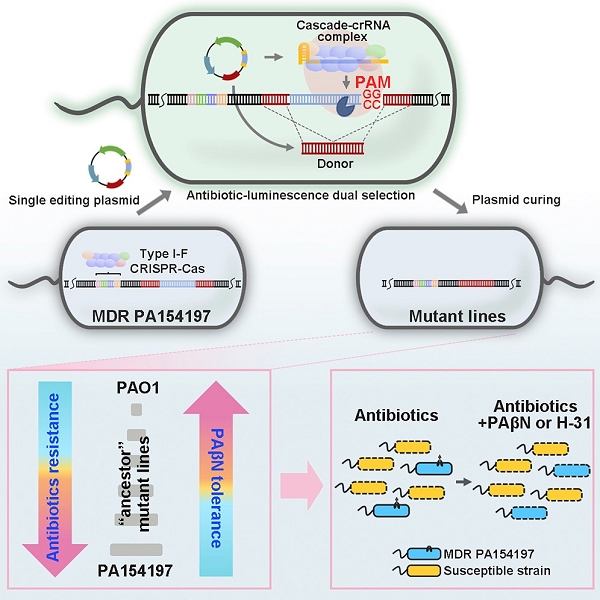
Antimicrobial resistance (AMR) is imposing an alarming threat on global public health. Particularly challenging are those "ESKAPE" pathogens that constitute the major sources of nosocomial infections with extraordinary drug resistance (i.e, Enterococcus spp, Staphylococcus aureus, Klebsiella spp, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp.). Among them, Pseudomonas aeruginosa is recognized as a prototypical multidrug-resistant (MDR) pathogen owing to both its intrinsic resistance to a variety of antimicrobials and its enormous capacity to develope acquired resistance during antibiotics chemotherapies (Oliver et al, 2015, Poole, 2011, Santajit and Indrawattana, 2016, Stover et al, 2000). Remarkably, in recent years, MDR international high-risk clones of P. aeruginosa have emerged and caused worldwide outbreaks (Oliver et al, 2015). Hence, it is of paramount importance to deeply understand the resistance mechanisms of these clones and develop anti-resistance treatment strategies. However, these studies are frequently impeded because of the lack of genetic-editing tools in the clinical P. aeruginosa genotypes.
P. aeruginosa is a ubiquitous pathogen capable of causing a wide array of acute and chronic infections. The species is notorious for its unusually large (ca. 6.4 Mbp) genome and extraordinarily diverse genotypes (Silby et al, 2011). As a result, genetic tools established in the model strains, such as PAO1 and PA14, are often inapplicable in the clinical, environmental, and biotechnological strains of interest. Moreover, clinical MDR clones often contain a complex set of resistance markers originating from both chromosomal gene mutations and transferable elements (Cabot et al, 2012, Hocquet et al, 2003), further limiting the conventional antibiotic-resistance-marker-based genetic manipulations. On the other hand, studies have indicated that the genetic background of resistant strains and epistasis among different resistant mutations plays an important role in the resistance levels of the strains and affects the effectiveness of antibiotic chemotherapies (Moura de Sousa et al, 2017, Vogwill et al, 2014, Vogwill et al, 2016). Hence, it is highly desirable to develop genome-editing techniques compatible with the genotypes of the strains of interests and to characterize AMR in the native genetic background of clinical strains.
In addition to being recognized as a prototypical MDR pathogen, P. aeruginosa is also an important model organism for understating CRISPR-Cas functions, especially the most widespread type I CRISPR-Cas system (van Belkum et al, 2015). To exert its functions in the adaptive immune process in bacteria, the Cas proteins specifically bind to and cleave the invader DNA based on Watson-Crick base pairing between a small CRISPR RNA (crRNA) and its DNA target (Barrangou et al, 2007, Hille et al, 2018). Conceivably, by modifying the sequence of the RNA guide, the Cas nucleases can be reprogrammed to cleave a specific genomic site and be exploited for genome editing upon a repair template is provided (Jinek et al, 2012). Phylogenetic analysis has revealed that CRISPR-Cas systems are widely distributed in global AMR P. aeruginosa isolates with ∼70% of them belonging to the I-F subtype (van Belkum et al, 2015). In recent years, repurposing the broadly distributed native CRISPR-Cas systems is emerging as a new CRISPR-based genome-editing strategy in prokaryotes. For instance, the native type I-B CRISPR-Cas system was harnessed for genome editing in the medically and industrially important species Clostridium tyrobutyricum and Clostridium pasteurianum, which frequently suffer from a poor DNA homeostasis and have a low genome-editing efficiency using the traditional allelic exchange or the heterologous CRISPR-Cas9 system (Pyne et al, 2016, Zhang et al, 2018). The endogenous type I-E CRISPR-Cas system was successfully repurposed for genome editing in the commensal bacterium Lactobacillus crispatus, which was formerly genetically recalcitrant (Hidalgo-Cantabrana et al, 2019). However, there has been no report about exploitation of native CRISPR-Cas systems for genome editing and functional genomics in the prototypic pathogen P. aeruginosa.
Previously, we have isolated an epidemic MDR P. aeruginosa clinical strain, PA154197, that shares a clonal complex with two international cystic fibrosis (CF) isolates and displays a comparable resistance profile to the high-risk clone ST175 (Cao et al, 2019). Employing the allelic exchange and the heterologous CRISPR-Cas9-based method failed to deliver desired mutations for resistance characterizations in this strain. As in the majority of clinical and environmental isolates of P. aeruginosa, PA154197 is found to contain a native type I-F CRISPR-Cas loci. Hence, in this study, we use PA154197 as a model to explore the native type I-F CRISPR-Cas-based genome editing in a clinical MDR P. aeruginosa genotype and its exploitation in the functional genomics of MDR. A highly efficient, one-step (for non-essential gene allele) or two-step insert-delete (In-Del) (for essential gene allele or inefficiently targeted genome site) genome-editing technique was successfully developed. The editing pipeline was readily applicable in additional clinical and environmental P. aeruginosa strains tested that contain the endogenous type I-F CRISPR-Cas system. Employing the technique, we unveiled the extraordinary resistance capacity of PA154197 and developed an advanced treatment strategy against this clinical MDR isolate. These studies provided a framework for understanding and control of clinical resistant pathogens.
Ubigene Biosciences is co-founded by biological academics and elites from China, the United States, and France. We are located in Guangzhou Science City, which serves as a global center for high technology and innovation. Ubigene Biosciences has 1000㎡ office areas and laboratories, involving genome editing, cell biology technology, and zebrafish research. We provide products and services for plasmids, viruses, cells, and zebrafish. We aim to provide customers with better gene-editing tools for cell or animal research.
We developed CRISPR-U™ and CRISPR-B™(based on CRISPR/Cas9 technology) which is more efficient than general CRISPR/Cas9 in double-strand breaking, CRISPR-U™ and CRISPR-B™ can greatly improve the efficiency of homologous recombination, easily achieve knockout (KO), point mutation (PM) and knockin (KI) in vitro and in vivo.
Genome Editing Platform
——Focusing on the Application of CRISPR-U™ and CRISPR-B™ Gene Editing Technology
Cell Biology Platform
——Focusing on primary cell
2. Provides culture strategies and related products for different cell types.3. Provides cell biology-related services such as cell isolation, extraction and validation.






 Phone
Phone
 Contact
Contact


